















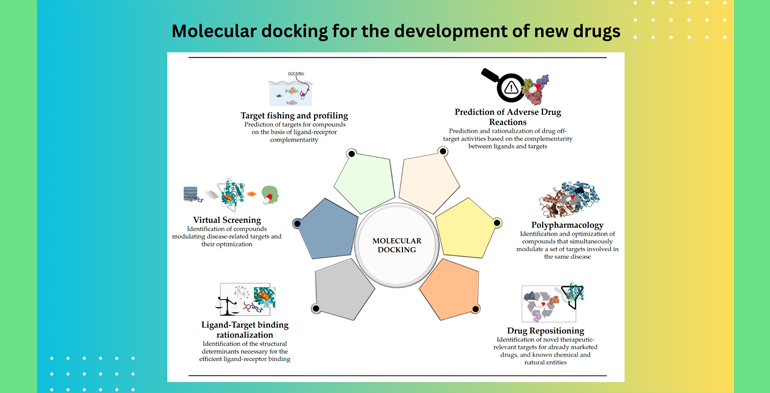
Dr. Md. Monirul Islam: Molecular docking is a computational technique widely used in drug discovery and design. It involves the prediction of how two or more molecules, typically a drug candidate and a target protein (usually a receptor or enzyme), interact with each other at the molecular level. The main goal of molecular docking is to predict the binding affinity and binding mode of a small molecule (the drug candidate) with a target protein. Here are the key steps and concepts involved in molecular docking for drug discovery:
1. Target Selection: The first step is to identify and select a specific target protein that plays a crucial role in a disease or biological process. This target protein is often chosen based on its known involvement in the disease or its potential as a therapeutic target.
2. Ligand Preparation: The drug candidate (ligand) needs to be prepared for docking. This includes determining its 3D structure, optimizing its geometry, and generating various conformations if needed.
3. Protein Preparation: The target protein structure is obtained, typically from experimental sources like X-ray crystallography or NMR spectroscopy. The protein structure may need preprocessing, such as adding missing atoms, assigning charges, and minimizing energy.
4. Grid Generation: A 3D grid or scoring function is generated around the binding site of the target protein. This grid defines the search space where the ligand will be docked.
5. Docking Algorithm: There are various docking algorithms available, each with its own approach. Common docking algorithms include AutoDock, AutoDock Vina, GOLD, and DOCK, among others. These algorithms use mathematical models and scoring functions to predict how the ligand will fit into the binding site of the protein.
6. Scoring Function: Docking algorithms rely on scoring functions to evaluate the binding affinity and predict the most energetically favorable binding mode. Scoring functions consider factors such as van der Waals forces, electrostatic interactions, hydrogen bonding, and solvation effects.
7. Ranking and Analysis: After performing multiple docking simulations, the ligand poses are ranked based on their predicted binding energies. Researchers analyze the top-ranked poses to select the most promising drug candidates.
8. Validation: Experimental validation is crucial to confirm the predicted binding affinity and binding mode of the drug candidate. Techniques like X-ray crystallography, NMR spectroscopy, or binding assays can be used for validation.
9. Lead Optimization: Once a promising drug candidate is identified, medicinal chemistry techniques are employed to optimize its properties for improved binding affinity, selectivity, and pharmacokinetics.
10. ADME/Toxicity Assessment: In parallel, the drug candidate's pharmacokinetic properties (absorption, distribution, metabolism, excretion) and potential toxicity are evaluated through in vitro and in vivo studies.
Molecular docking is a valuable tool in the early stages of drug discovery, as it helps narrow down the pool of potential drug candidates, saving time and resources before moving on to more expensive and time-consuming experimental testing. However, it's important to note that molecular docking predictions are computational approximations and require experimental validation to confirm their accuracy and reliability.
-Writer: Senior Scientist, ASRBC, ACI Seed


